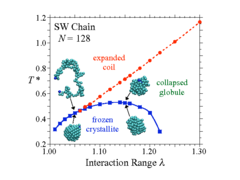
 A single polymer chain in a poor solvent may collapse into a dense fluid globule, but it may instead also crystallize: By extensive simulations with the Wang-Landau algorithm we have shown that the crystal is favorable if the range of the attractive interactions between the monomers exceeds the range of the repulsions only slightly. These findings may be useful to understand scenarios for protein crystallization. The resulting structure is also modified when an attractive substrate surface is present, and/or when one considers a semiflexible rather than a flexible polymer: then liquid-crystal-line ordering comes into play. Single chains then may collapse forming torodial or plate-like strucutures, or lamellae attached to walls. Multichain systems, or semi-flexible polymers, however, are found to undergo isotropic to nematic transitions, similar to systems of hard rods. In the presence of confinement into thin films by hard walls, "capillary normalization" (i.e. wall-induced nematic order) is found. This research is carried out in collaboration with V. A. Ivanov (Moscow State University), J. Luettmer-Strathmann (The University of Akron), M. P. Taylor (Hiram College), and W. Paul (Martin Luther Universität Halle.) For more Information, please contact Kurt Binder . A single polymer chain in a poor solvent may collapse into a dense fluid globule, but it may instead also crystallize: By extensive simulations with the Wang-Landau algorithm we have shown that the crystal is favorable if the range of the attractive interactions between the monomers exceeds the range of the repulsions only slightly. These findings may be useful to understand scenarios for protein crystallization. The resulting structure is also modified when an attractive substrate surface is present, and/or when one considers a semiflexible rather than a flexible polymer: then liquid-crystal-line ordering comes into play. Single chains then may collapse forming torodial or plate-like strucutures, or lamellae attached to walls. Multichain systems, or semi-flexible polymers, however, are found to undergo isotropic to nematic transitions, similar to systems of hard rods. In the presence of confinement into thin films by hard walls, "capillary normalization" (i.e. wall-induced nematic order) is found. This research is carried out in collaboration with V. A. Ivanov (Moscow State University), J. Luettmer-Strathmann (The University of Akron), M. P. Taylor (Hiram College), and W. Paul (Martin Luther Universität Halle.) For more Information, please contact Kurt Binder .
|
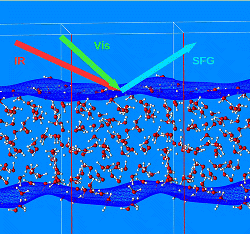 Confined water at nanoscale shows properties which are remarkably different from bulk. Vibrational Sum Frequency Generation Spectroscopy (VSFG) has contributed to a large extent to draw the attention on the new physical and chemical properties at interfaces, thanks to its ability to selectively probe non-centrosymmetric systems. However, the microscopic characterization of the VSFG spectra remains hard even with the development of some improvements like Phase-Sensitive VSFG. Molecular dynamics simulations can play a key role to provide a molecular interpretation of the spectra. In particular DFT-based simulations which do not require a priori parametrization, are particularly suitable to address the heterogeneous environment at interfaces. Our aim is to advance computational spectrscopy methods in order to provide a microscopic understanding of the special water structure and dynamics at interfaces. For more information, please contact
Confined water at nanoscale shows properties which are remarkably different from bulk. Vibrational Sum Frequency Generation Spectroscopy (VSFG) has contributed to a large extent to draw the attention on the new physical and chemical properties at interfaces, thanks to its ability to selectively probe non-centrosymmetric systems. However, the microscopic characterization of the VSFG spectra remains hard even with the development of some improvements like Phase-Sensitive VSFG. Molecular dynamics simulations can play a key role to provide a molecular interpretation of the spectra. In particular DFT-based simulations which do not require a priori parametrization, are particularly suitable to address the heterogeneous environment at interfaces. Our aim is to advance computational spectrscopy methods in order to provide a microscopic understanding of the special water structure and dynamics at interfaces. For more information, please contact 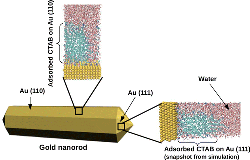 We investigate how the properties at the interface influence the crystal growth in a few selected example. In particular one topic is related to the understanding of the microscopic origin of the asymmetric growth mechanism in gold nanorods (Collaborators: C. Sönnichsen, Chemistry, JGU). The second topic is related to understanding how bio-polymers such as polyacrylate, poly-aspartate and poly-glutamate influence the crystalline phase, morphology and growth rate of calcium oxalate. Ab initio molecular dynamics study of the interactions of the water/mineral and water/polymer/mineral interfaces shed light on the biomineralization process and on the mechanisms responsible for its inhibition. For more information, please contact
We investigate how the properties at the interface influence the crystal growth in a few selected example. In particular one topic is related to the understanding of the microscopic origin of the asymmetric growth mechanism in gold nanorods (Collaborators: C. Sönnichsen, Chemistry, JGU). The second topic is related to understanding how bio-polymers such as polyacrylate, poly-aspartate and poly-glutamate influence the crystalline phase, morphology and growth rate of calcium oxalate. Ab initio molecular dynamics study of the interactions of the water/mineral and water/polymer/mineral interfaces shed light on the biomineralization process and on the mechanisms responsible for its inhibition. For more information, please contact 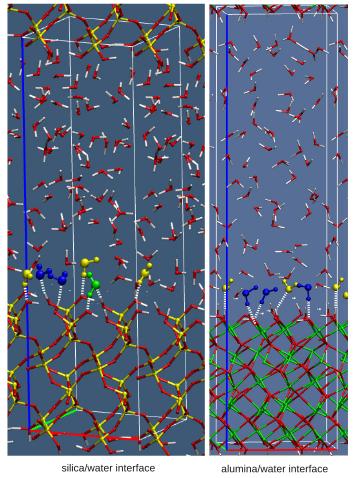 We aim to a detailed understanding of the molecular behaviour of the different solid–water interfaces, using density functional theory based molecular dynamics (DFTMD) simulations, where a consistent treatment of the electronic structure of solvent and surface is provided. Our interest includes oxide water interfaces (such as silica, alumina, clays) as well as ionic salts/water interfaces, such as the fluoride/water interface. For more information, please contact
We aim to a detailed understanding of the molecular behaviour of the different solid–water interfaces, using density functional theory based molecular dynamics (DFTMD) simulations, where a consistent treatment of the electronic structure of solvent and surface is provided. Our interest includes oxide water interfaces (such as silica, alumina, clays) as well as ionic salts/water interfaces, such as the fluoride/water interface. For more information, please contact